Results¶
folders¶
Basically, there are 4 folders, including 0-shells, 1-data, 2-results, 3-reports, and 4-logs. Their introduction is as following tables(same as the table in Storage section, 600 genome with 100x data):
| Folder | Storage | Description |
|---|---|---|
| 0-shells | 1.8M | Store all the shells |
| 1-data | 61G | Store the filted fasta file and as input for mecat |
| 2-results | 9.9G | Store the mecat results, picked fastq file and PBJelly results |
| 3-reports | 5.5K | Store reports for run_pbjelly (just a txt file now) |
| 4-logs | 340G | Store log file for run_pbjelly and tmp file for mecat alignement(Count for the vast majority) |
Scaffold file¶
After filling the gaps, run_pbjelly output all the sequences in the file named ‘jelly.out.fasta’ under the file folder ‘2-results’.
Gap informations¶
Besides, for the detailed process information for each gap, you can refer the file ‘gap_fill_status.txt’ also under the file folder ‘2-results’.
Reports¶
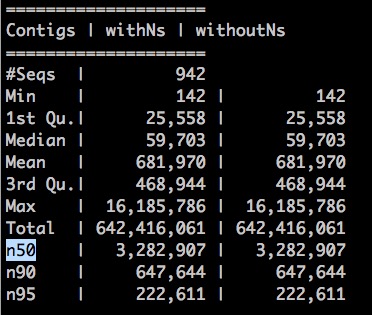
We also generated a html report for run_pbjelly, which include contig N50 value, distribution of gap lengths, and et al. And the report files were placed under the file folder ‘3-reports’ (comming soon). A simple statistics were generated under the file folder ‘3-reports’, such as: